
The U.S. Food and Drug Administration doesn’t just approve new drugs-it keeps millions of Americans alive by making sure affordable generic versions are safe, effective, and available. Every day, generic drugs make up 9 out of 10 prescriptions filled in the United States. But behind that number is a complex, science-driven system designed to ensure that a $5 generic pill works just as well as its $300 brand-name counterpart. This isn’t guesswork. It’s regulation. And it all runs through the FDA’s Office of Generic Drugs.
How Generic Drugs Get Approved: The ANDA Pathway
Generic drugs don’t go through the same approval process as new drugs. That’s by design. When a brand-name drug is first developed, the company spends over $2 billion and a decade or more running clinical trials to prove it’s safe and works. But once the patent expires, other companies can make the same drug-without repeating those expensive trials. The FDA created the Abbreviated New Drug Application, or ANDA, to make this possible.
An ANDA doesn’t need to prove the drug works. It only needs to prove it’s the same. That means identical active ingredients, same strength, same form (pill, injection, cream), same way it’s taken (oral, topical, etc.), and same medical use. The only things allowed to be different? The inactive ingredients-like dyes or fillers-that don’t affect how the drug works in your body.
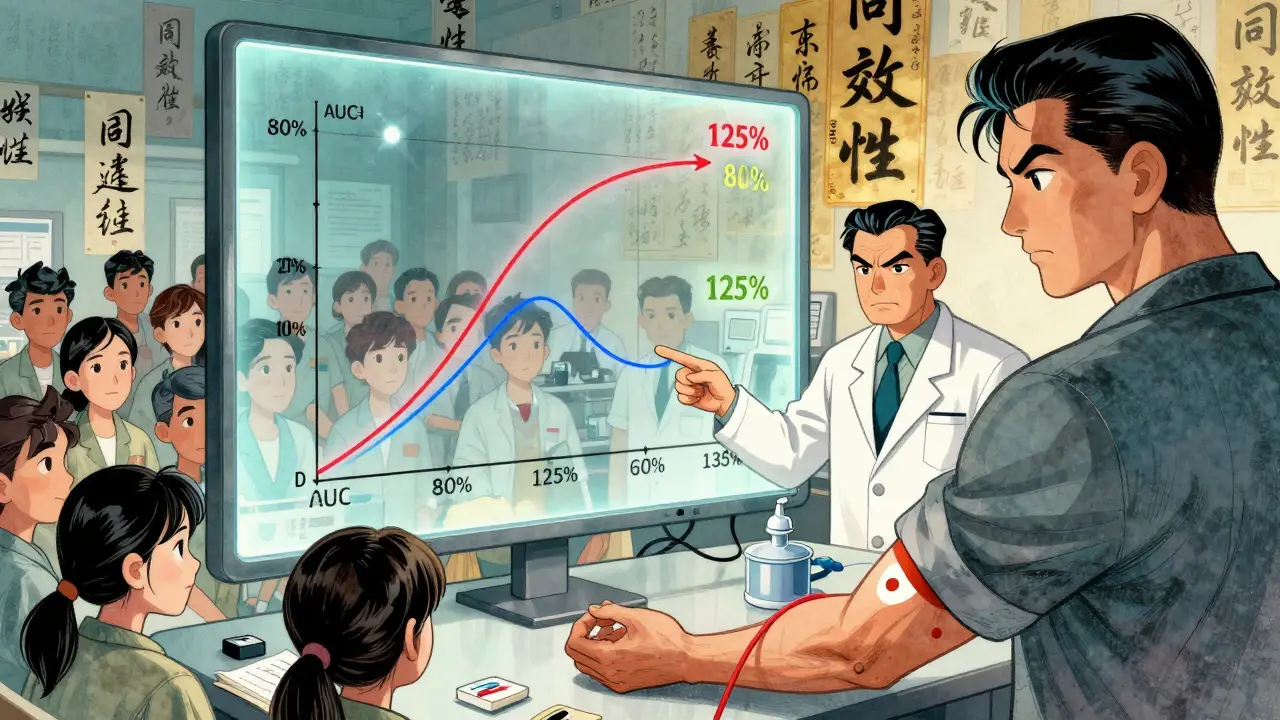
The real test? Bioequivalence. The FDA requires generic manufacturers to show that their version is absorbed into the bloodstream at the same rate and to the same extent as the brand-name drug. This is done with studies on 24 to 36 healthy volunteers. Blood samples are taken over time to measure two key numbers: AUC (how much of the drug gets into your system) and Cmax (how fast it gets there). The FDA’s rule? The generic’s results must fall between 80% and 125% of the brand’s. If it’s outside that range, the application gets rejected.
What Happens After the Application Is Submitted
Submitting an ANDA isn’t like mailing a form. It’s like sending a 15,000- to 20,000-page scientific dossier. Every detail matters: how the drug is made, how the ingredients are tested, how the factory is cleaned, even how the tablets are pressed. The FDA’s Division of Filing Review checks for completeness first. In 2022, 15.3% of applications were refused outright-called Refuse-to-Receive, or RTR-because they were missing key information, especially in the chemistry and manufacturing sections.
If it passes filing, it moves to substantive review. The FDA has set deadlines under the Generic Drug User Fee Amendments (GDUFA). Standard applications get a 10-month review window. Priority applications-like first generics or drugs in short supply-get 8 months. In 2023, the FDA approved 1,256 ANDAs, up from 1,116 the year before. But there were also 317 RTR decisions. That’s not failure. It’s quality control.
Manufacturing facilities aren’t left out. Every plant making generic drugs must follow Current Good Manufacturing Practices (CGMP). The FDA inspects these facilities regularly. In 2023, 82.7% of generic drug sites were inspected. If one fails, the entire product line can be blocked-even if the science is perfect.
Why This System Saves Billions
Generic drugs aren’t just cheaper-they’re transformative. In 2023, they saved the U.S. healthcare system $132.6 billion. That’s not a guess. It’s from the Generic Pharmaceutical Association’s official report. The average patient pays 80-85% less for a generic than a brand-name drug. A patient on insulin might drop from $390 a month to $98 after the FDA approves a generic version. That’s life-changing.
But the savings aren’t just for patients. Employers, insurers, Medicare, and Medicaid all benefit. Generics make up only 23% of total drug spending, but they account for 90% of prescriptions. That’s the power of scale and regulation working together.
Complex Generics Are the New Frontier
Not all generics are created equal. Simple pills? Easy. Inhalers? Creams? Extended-release tablets? Those are complex. They don’t just need the same ingredients-they need the same delivery system. A generic inhaler must spray the exact same amount of medicine into the lungs at the same speed. A cream must penetrate the skin the same way. These aren’t just chemistry problems-they’re engineering challenges.
In 2018, only 22.1% of approved generics were complex. By 2023, that number jumped to 37.5%. The FDA has made this a priority. Their Complex Generic Drugs Initiative includes new guidance, better testing methods, and faster review paths. In 2024, they started piloting AI tools to help reviewers analyze data faster. By 2026, they aim to use real-world evidence-like patient data from electronic health records-to support approvals for some complex products.
But even with progress, it’s hard. Only 83.6% of first generics were approved in 2023, up from 67.2% in 2018. The gap is closing, but it’s still a bottleneck.
What Goes Wrong-and What Doesn’t
People worry: Are generics safe? Do they work the same? The FDA tracks this closely. Between 2020 and 2023, there were 1,485 adverse event reports linked to generics in the agency’s database. But when the FDA investigated, 92.3% of those cases were due to the patient’s underlying condition getting worse-not the drug itself.
Surveys back this up. In 2023, CVS Health found 78.4% of patients trusted FDA-approved generics. 63.2% said they noticed no difference in effectiveness compared to the brand-name version they used before.
Still, complaints exist. Some patients report feeling different on a generic. Pharmacists hear it all the time. But when you look at the science, those differences rarely come from the drug. They come from expectations. Or from switching between multiple generic brands that, while each meets FDA standards, have slightly different inactive ingredients that affect how the pill feels or tastes.
The Cost of Approval-and Who Pays
Applying for an ANDA isn’t free. Under GDUFA III, which took effect in October 2022, the FDA charges $389,490 per application. Facilities pay between $207,700 and $415,400 a year in fees, depending on size and type. These aren’t taxes. They’re user fees-paid by the companies, not taxpayers. That money funds the review teams, inspections, and technology upgrades.
Companies that succeed usually have teams of 8 to 12 specialists: regulatory experts, pharmacologists, chemists, and quality control engineers. First-time applicants often take 18 to 24 months to get their first ANDA right. Common mistakes? Incomplete manufacturing details (41.7% of RTRs) and poorly designed bioequivalence studies (28.3% of RTRs).
Successful applicants use pre-ANDA meetings with the FDA. In 2022, 78.4% of approved applications had one. These meetings let companies ask questions before spending millions on studies. It’s like getting feedback before submitting a thesis.
How the U.S. Compares to the Rest of the World
The U.S. system is unique. In Europe, the EMA sometimes requires additional clinical data-even for simple generics. In Japan, every single generic must undergo in vivo bioequivalence testing, no matter how simple the drug. The U.S. system is faster, more efficient, and more predictable.
But the U.S. isn’t perfect. In 2022, Senator Bernie Sanders’ committee found 1,842 ANDAs were still pending, with 317 waiting over three years. The FDA admitted that 14.8% of applications in 2022 got complete response letters because of bioequivalence issues. Resources are stretched thin.
That’s why GDUFA IV, agreed on in September 2024, committed $2.1 billion over three years-including $412 million specifically for complex generics. It’s also why the FDA launched a new pilot in October 2025: faster reviews for companies making generics in the U.S. Their goal? Cut review times by 30% for those applicants.
What’s Next for Generic Drug Approval
The pipeline is full. As of Q1 2024, there were 2,147 pending applications for first generics-drugs that have never been copied before. That’s a record. The FDA expects to approve 1,500 to 1,700 ANDAs annually by 2027.
The biggest challenges ahead? Supply chain risks-78% of active ingredients come from outside the U.S.-and patent litigation. On average, each brand-name drug facing generic competition gets hit with 34.7 patent challenges. The FDA’s Drug Competition Action Plan has already cut first-generic approval times by 37.2% since 2017. But the pressure is growing.
One thing won’t change: the FDA’s job. They don’t approve drugs to save money. They approve them to save lives. And with 90% of prescriptions filled with generics, that job is bigger than ever.
Nancy Kou
December 19, 2025 AT 17:29The FDA's generic drug approval system is one of the most underappreciated public health triumphs in modern medicine. It's not just about cost-it's about access. Millions of diabetics, hypertensives, and psychiatric patients rely on these pills to survive, and without the ANDA pathway, many would be priced out of treatment entirely. The bioequivalence standards are brutal but necessary. 80-125% isn't arbitrary-it's the sweet spot between safety and practicality. And yes, the inspections are relentless, and yes, 15% of applications get rejected before review even starts. That's not bureaucracy. That's vigilance.
Jedidiah Massey
December 20, 2025 AT 04:09Let’s be real-the ANDA pathway is a masterclass in regulatory arbitrage. You’re essentially leveraging the original innovator’s R&D spend while avoiding the burden of clinical validation. The bioequivalence metrics? A statistical fiction. Cmax and AUC don’t capture pharmacodynamic variability across polymorphic metabolizers. And don’t get me started on the CGMP inspections-those are theater. Factories in India and China pass with flying colors, then ship substandard API’s that degrade in transit. The FDA’s metrics are optimized for throughput, not true therapeutic equivalence.
Takeysha Turnquest
December 20, 2025 AT 15:51Generics save billions but they don’t save souls. The real tragedy isn’t the price tag-it’s the quiet erosion of trust. People switch from brand to generic and feel weird. Not because the drug is different-but because they were told it’s the same. And when you’re on lithium or warfarin or thyroid meds, ‘the same’ isn’t enough. You need the same in your blood, your nerves, your bones. The FDA’s 80-125% range is a compromise. Not a guarantee. We treat medicine like a commodity. It’s not. It’s biology.
Hussien SLeiman
December 22, 2025 AT 04:23Everyone acts like the FDA is some infallible guardian of public health but let’s cut through the PR. The agency approves over a thousand ANDAs a year and yet still has 1,800 pending applications sitting in limbo for years. That’s not efficiency-that’s dysfunction. And the user fees? Pure corporate welfare. Companies pay $389K to submit an application and then get a 10-month review clock that’s constantly extended by their own sloppy submissions. The whole system is a feedback loop of incompetence disguised as regulation. If you want real reform, abolish GDUFA and let the market sort it out. Let pharmacists and doctors choose. Let patients vote with their wallets. The FDA doesn’t make drugs better-it just makes them slower and more expensive to bring to market.
Emily P
December 22, 2025 AT 15:39I’ve been on a generic version of sertraline for five years. I switched from the brand after my insurance changed. I didn’t notice a difference. But I also didn’t know what to look for. What would ‘different’ even feel like? I wonder how many people report side effects because they expect to, not because they’re real. The science says it’s equivalent. But the human experience? That’s a different dataset. I wish the FDA published more patient-reported outcomes alongside the bioequivalence data.
Vicki Belcher
December 23, 2025 AT 19:09Y’all need to chill. 🙏 The FDA is doing the heavy lifting so you don’t have to pay $400 for your blood pressure med. 💊 I’ve seen friends choose between insulin and rent. Generics aren’t perfect-but they’re the reason someone’s alive today. The system’s flawed? Sure. But it’s saving billions and lives. Let’s fix it, not trash it. 🌟
Nicole Rutherford
December 25, 2025 AT 03:37Of course the FDA approves 1,256 ANDAs a year-because they’re under pressure from Big Pharma to keep generics flowing so the brand names can raise prices on the few remaining patents. The ‘complex generics’ push? A distraction. They’re not investing in innovation-they’re investing in delay. The 83.6% approval rate for first generics? That’s not progress. That’s the system grinding to a halt under its own weight. And the inspections? A joke. Plants with multiple violations still get approved. You think they’re checking the air quality in the vial sterilization room? No. They’re checking the paperwork. Again.
Allison Pannabekcer
December 26, 2025 AT 03:44Let’s not forget who’s really behind this: the patients. The single mom on insulin. The veteran on warfarin. The elderly man with congestive heart failure. They don’t care about GDUFA or Cmax or bioequivalence percentages. They care if they can afford to live. The FDA’s job isn’t to be perfect-it’s to be practical. The system isn’t flawless, but it’s the best we’ve got. And before you trash it, ask yourself: would you rather have a $5 pill that works 98% of the time, or a $300 pill that only a few can afford? The answer’s obvious. Let’s keep improving it-not tearing it down.